

There is increasing acceptance of the burden of non-motor symptoms (NMS) and their importance in our effective and personalized management of PD. NMS are part of the heterogeneity of the disease and – along with genetics, imaging and the measurement of markers such as α-synuclein in body tissues and fluids — may help identify people at high risk of developing PD and those who are already in a prodromal phase. The wide range of NMS associated with PD and its prodrome supports the view that the disease involves multiple anatomical sites and abnormalities of neurotransmission.1,2,3
NMS and the complex pathology of PD
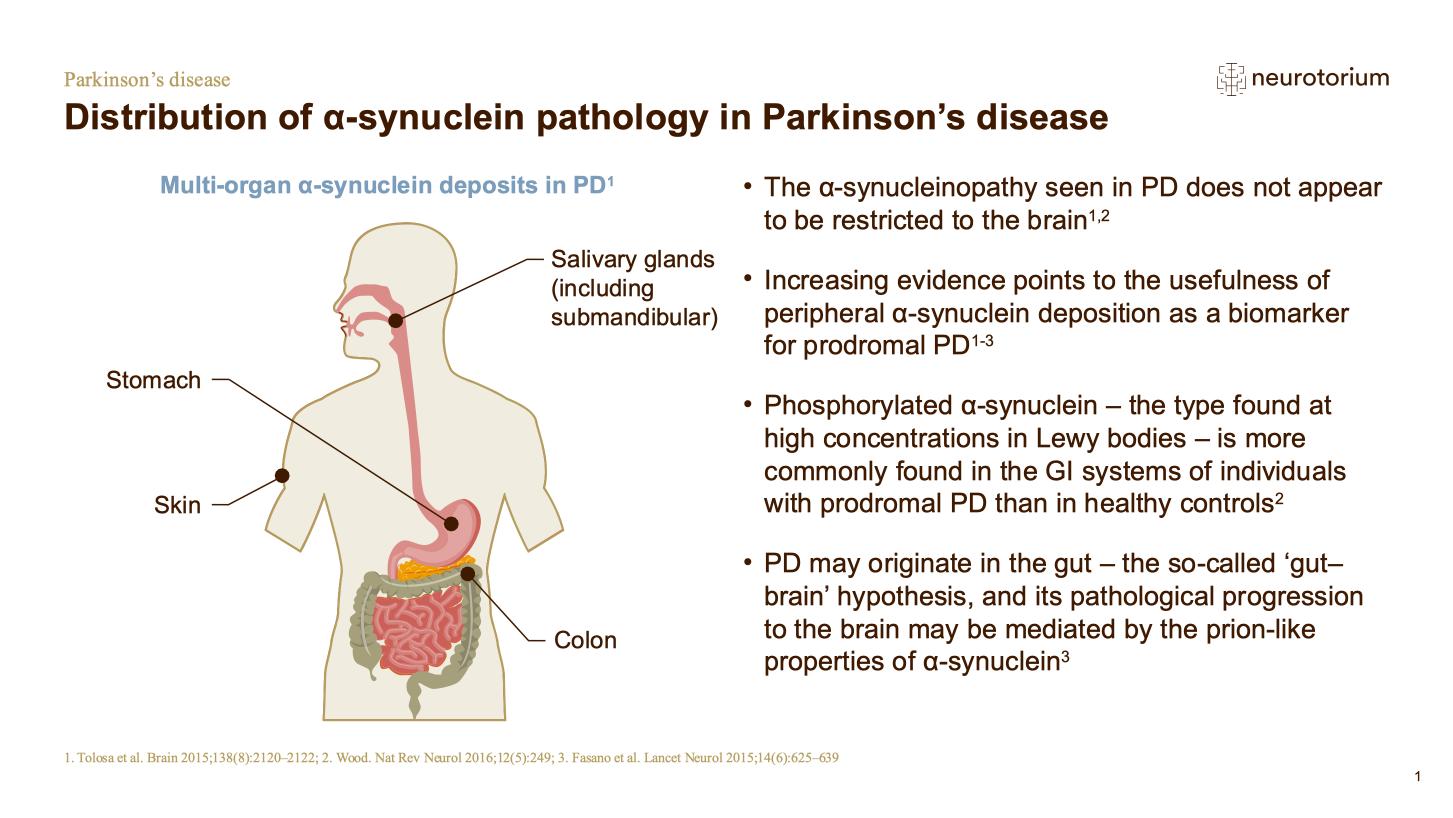
Distribution and Spread of Abnormal α-synuclein
PD is associated with a body-wide distribution of α-synuclein aggregates, which are found, for example, in the olfactory bulbs, colon, heart and skin (Fig 1). Involvement of the autonomic nervous system, with accompanying autonomic dysfunction such as constipation, appears to be an early phase in disease development.1
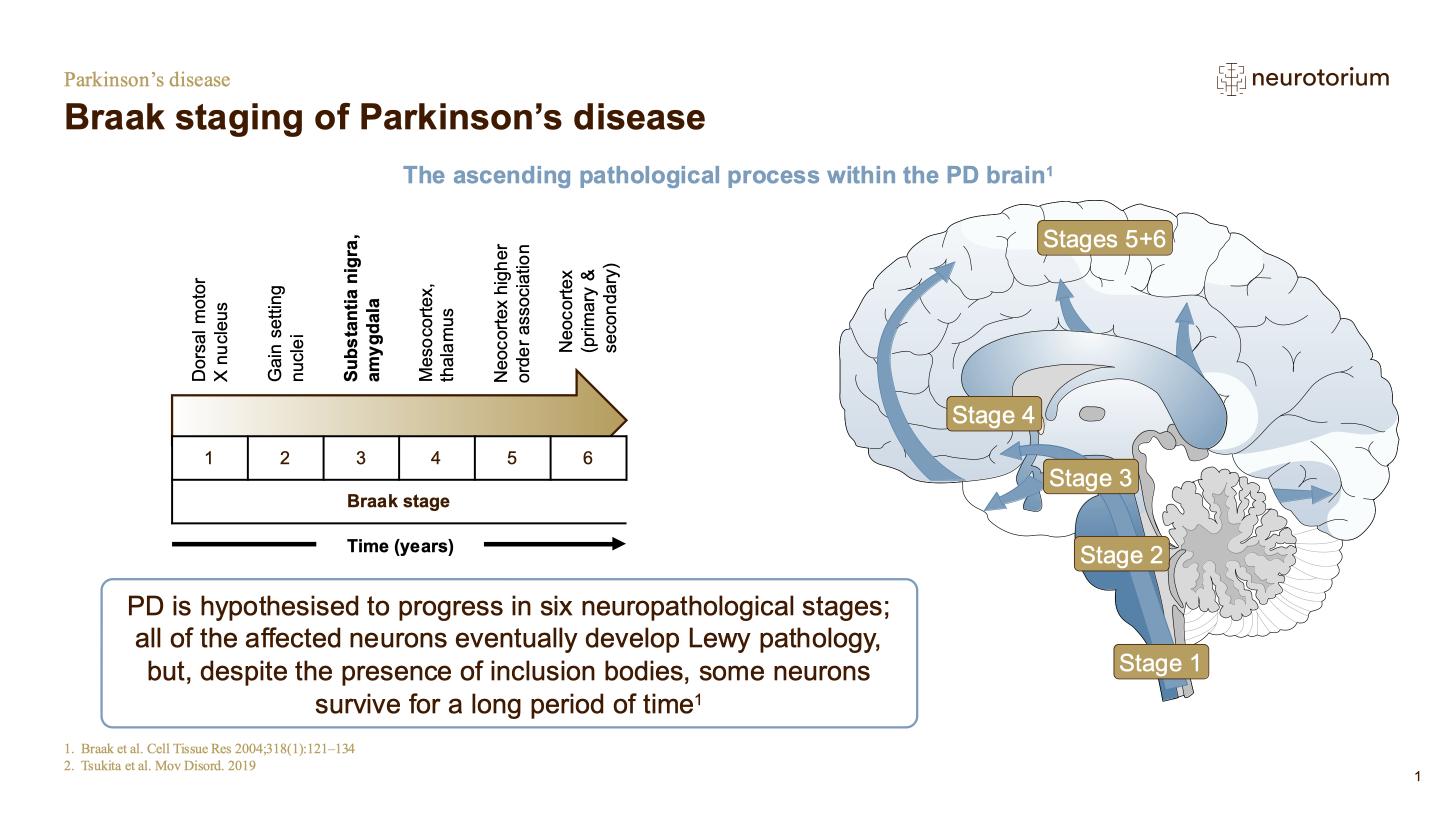
The presence of NMS in the prodromal phase of PD supports the Braak hypothesis that Lewy pathology develops in the dorsal motor nucleus of the vagus nerve before spreading subsequently to the substantia nigra, areas of the midbrain and basal forebrain, and finally the neocortex (Fig 2). The sequence of NMS is compatible with this model, with hyposmia and sleep disturbance accompanying early involvement of olfactory centres and lower brainstem, and cognitive and psychiatric symptoms emerging later as cortical structures are implicated.
Involvement of multiple neurotransmitter systems
The classical motor signs and symptoms of PD are clearly associated with loss of nigral dopaminergic neurons. However, dopamine is not the only neurotransmitter deficiency involved in this multi-faceted disorder. Nataliya Titova et al have recently argued that the speed and extent of loss of non-dopaminergic neurons may actually be greater than that of dopaminergic neurons, at least in the prodromal and early stages of the PD syndrome.4
Levels of norepinephrine (NE) are abnormally low in several areas of the central nervous system and in the peripheral sympathetic neurons of people with PD; and NE deficiency can be linked to non-motor features of the disease such as orthostatic hypotension, sleep disturbance, depression, apathy and impaired memory and attention.5 Alberto Espay and colleagues describe the extensive loss of noradrenergic neurons in the locus coeruleus of the PD brain and the consequent reduction in noradrenergic projections to structures from the hippocampus to the frontal cortex. Based on this pathology, they argue that greater attention should be paid to NE and the potential clinical benefits of its enhancement.
Cholinergic denervation has also been implicated; and degeneration of the forebrain cholinergic system appears early in the disease course.4 In a recent review of four studies, each of which used stereological counting methods, the average loss of cholinergic neurons in the pedunculopontine nucleus of patients with PD was 41%.6 Loss of central cholinergic function has been related to cognitive symptoms, from mild impairment to dementia, while reduced peripheral cholinergic activity has been associated, for example, with gastrointestinal dysfunction.4
Serotonergic neurons in the raphe nuclei innervate the hippocampus, hypothalamus, and several regions of the cortex. Lewy pathology in the raphe nucleus is seen early in PD, along with loss of serotonergic neurons.7 The extent of this loss has been associated with depression, reinforcing the idea that deficits in serotonin systems are implicated in PD-related mood disorders. Serotonergic deficits may also contribute to cognitive impairment.
“Loss of central cholinergic function has been related to cognitive symptoms, from mild impairment to dementia”
NMS are a central feature of the PD prodrome
Although not the most common prodromal feature, REM Sleep Behavior Disorder (RBD) is the NMS that is most predictive of subsequent PD. In a major review to mark two hundred years since James Parkinson’s celebrated Essay, Obeso et al compared prodromal markers.8 The approximate relative risk (RR) assigned to RBD was 50. This compared with an RR of 5 for olfactory disorder, and of 2.5 for constipation.
Also striking is the length of time by which RBD and other NMS can precede PD, which may not be diagnosed for several decades after appearance of the non-motor symptoms.
In a Mayo clinic series of 27 patients who had idiopathic RBD and subsequently developed a neurodegenerative syndrome – PD, Multiple System Atrophy (MSA), or Dementia with Lewy Bodies (DLB) – at least fifteen years later, the median interval between these events was 25 years and the maximum interval was fifty.9
Using linked records from the Rochester Epidemiology Project in Minnesota, USA, Savica and colleagues at the Mayo Clinic established that constipation preceded the onset of PD (or the index year) significantly more frequently in cases than in matched controls (Odds Ratio 2.48).10 The association was independent of smoking and caffeine consumption. Intriguingly, it remained significant even when analysis was confined to constipation more than twenty years prior to the onset of motor symptoms.
In a larger case-control study (overall n=8166 cases; 46,755 controls) conducted in the UK, people who went on to develop PD were significantly more likely than controls to have had constipation five years before diagnosis (RR 2.24).11 Even ten years before diagnosis, the association was still significant (RR 2.01). Table 1 shows the RRs associated with other prodromal autonomic and neuropsychiatric features, along with that attributable to the presence five years before diagnosis of tremor itself.
“High relative risk and long latency make REM sleep behavior disorder a compelling target when selecting people for PD prevention studies”
Table 1: Conditions present significantly more frequently in PD cases than in controls five years before PD diagnosis
| Relative risk | ||
| Motor Features | Tremor | 13.7 |
| Impaired balance | 2.19 | |
| Autonomic Features | Constipation | 2.24 |
| Hypotension | 3.23 | |
| Erectile dysfunction | 1.30 | |
| Urinary dysfunction | 1.96 | |
| Dizziness | 1.99 | |
| Neuropsychiatric | Fatigue | 1.56 |
| Depression | 1.76 | |
| Anxiety | 1.41 |
Schrag et al. Lancet Neurology 201511. Based on primary care data from the Health Improvement Network UK. In this study, REM sleep behavior disorder and anosmia were reported in fewer than 1% of people per thousand person-years and were excluded from analysis.
Whether individual prodromal NMS can be grouped together to further enhance their predictive power, with or without the addition of other markers – including the presence of genes that enhance the risk of developing PD – is a pressing question considered below. So too is the potential of NMS in helping differentiate between subtypes of PD.
NMS and the subtyping of PD
Non-motor features as markers of risk and progression
Patients differ in the nature and severity of their NMS. In part, these differences are predictable on the basis of etiological factors. This is clear, for example, with certain glucocerebrosidase (GBA) polymorphisms and mutations that increase risk of dementia.
In a large Italian study, GBA mutation carriers were more likely than non-carriers to have severe motor disease, and also more likely to have dementia (HR=3.16).12 This was especially so for carriers of severe GBA mutations: Presence of the L444P variant was associated with a more aggressive phenotype akin to that of Dementia with Lewy Bodies. In a more recent study from Scandinavia, half of the 12% of incident PD patients who had GBA variants progressed to dementia within seven years.13 This rate of progression was faster than that seen in non-carriers.
The presence of one non-motor feature can also be predictive of the presence of others. Thus, PD patients with RBD are more likely than those without sleep disorder to have cognitive impairment.14 This led some investigators to think that PD with RBD might represent a distinct PD phenotype. Being able to identify a PD subtype (whether related to gene mutation or other factors) that conferred enhanced risk of cognitive decline would in principle be clinically useful, since cognitive training might be implemented at an early stage.15
In a prospective study of 113 people with PD (mean age 67 years) attending two Montreal movement disorder clinics, Fereshtehnejad et al used cluster analysis to group patients according to clinical features.16 Investigators were able to re-assess 76 patients after a median of 4.5 years.
Analysis suggested patients could be divided into three groups: those with mainly motor disease and slow progression; those with diffuse/malignant disease; and those with an intermediate phenotype and disease course. Patients in the diffuse/malignant group were more likely than the others to have had mild cognitive impairment, orthostatic hypotension and RBD at baseline.
This distinct phenotype was present despite similar age and disease duration, suggesting that it represented a true subgroup and not a more advanced stage of the same pathophysiological entity. Based on the evidence that this cluster of NMS indicated risk of rapid progression in both motor and non-motor symptoms, the authors recommended baseline screening of all PD patients for cognitive impairment, hypotension and sleep disorder.
“Studies of GBA mutation carriers show convincingly that PD risk genes can influence the severity of non-motor symptoms”
Examples of subtyping based on motor and non-motor features
The possibility that urinary symptoms might serve as a simple clinical marker of more rapid disease progression was suggested by Erro and colleagues.17 The median time from diagnosis to introduction of L-dopa was significantly shorter for patients with urinary problems (median 20 months) than those without this feature (37 months).
Sauerbier et al found a more complex picture in a study that identified seven distinct subtypes of PD characterized by the most dominant NMS present. Their analysis suggested subgroups in which the most salient symptoms were cognitive impairment, or apathy, or depression and anxiety, or sleep disturbance, or pain, or fatigue, or autonomic dysfunction.18 The authors further suggested that sleep-dominant and autonomic-dominant subtypes might be considered together as a phenotype deriving from underlying brainstem and olfactory system pathology, while the subtype dominated by cognitive problems represented a group suffering from mainly cortical involvement.
Another group of researchers cluster analyzed symptoms experienced by a large international cohort including 904 patients from the range of motor stages. The study identified four subtypes: those mildy affected in both motor and non-motor domains; those with severe NMS but mild motor symptoms; those with mild NMS but severe motor features; and those severely affected in both domains.19
This study used validated scales to assess NMS. However, a wide consensus on which scales should be used has yet to emerge. And the 2015 MDS diagnostic criteria made clear that at that stage we were not able to reliably identify subtypes of PD in a way that would be clinically useful.20
Indeed, as Marras and Chaudhuri point out, the subtyping project is likely to prove challenging since each disease-associated feature lies somewhere along a spectrum, and features are not likely to cluster in distinct, non-overlapping groups.21 The problem will be compounded if Alberto Espay et al are correct in arguing that PD is at least twenty different diseases.22 On this view, degeneration in the nigral dopamine system is common to a range of conditions, but these conditions differ in their molecular and genetic etiologies, and clinical pathology.
That said, there is considerable impetus in this direction, and much research effort is being directed towards the identification of subtype “signatures” that reflect a distinct pathophysiology relevant to prognosis and the much-needed individualization of management.23
“If PD is reached via several different pathological pathways, finding common markers of progression will be difficult”
NMS in the strategy to prevent PD
NMS have potential to contribute – along with other clinical, imaging and biofluid markers – to the identification of people in the prodromal phase of PD who are at high risk of developing classical motor symptoms. The ability to do this offers opportunities for early intervention, and perhaps prevention – assuming that we can develop neuroprotective, disease-modifying agents that are well tolerated.
The potential for such an approach is illustrated by the Parkinson Associated Risk Study (PARS) which showed that the combined presence of hyposmia and a deficit on dopamine transporter (DAT) imaging is highly predictive of conversion to PD within four years.24 At the outset of the study, olfactory screening in the community identified 203 people with hyposmia who were subsequently invited for DAT imaging. Among 21 subjects who were hyposmic and had a DAT deficit at baseline (65% or less of expected binding), 67% had been diagnosed with PD by four years. The RR compared with people with an intermediate or no baseline DAT deficit was 17.5.
The fact that idiopathic RBD is also a powerful predictor of synucleinopathies is being used to advantage in a parallel initiative.25 Data contributed by the 24 centers belonging to the international RBD Study Group were used to identify factors relevant to phenoconversion among people with sleep disorder at baseline. In this major study of 1280 subjects with RBD at the outset, the rate of conversion to PD, DLB or MSA was 74% at twelve years. Risk of phenoconversion was significantly increased by a range of motor and non-motor features (Table 2).
“Hyposmia plus DAT deficit is highly predictive of conversion to PD within 4 years; 24 RBD is highly predictive of synucleinopathy over 12 years25”
Table 2: Factors predicting conversion to PD, DLB or MSA in People with RBD followed for twelve years
| Hazard Ratio | |
| Abnormal Quantitative Motor Testing | 3.16 |
| Objective Motor Examination | 3.03 |
| Olfactory Deficit | 2.62 |
| Mild Cognitive Impairment | 1.91-2.37 |
| Erectile Dysfunction | 2.13 |
| Motor Symptoms | 2.11 |
| Abnormal DAT Scan | 1.98 |
| Colour Vision Abnormalities | 1.69 |
| Constipation | 1.67 |
| REM Atonia Loss | 1.54 |
Postuma et al 201925. In this study, age (HR = 1.54) also predicted phenoconversion. There was no significant effect of sex, daytime somnolence, insomnia, restless legs syndrome, sleep apnea, urinary dysfunction, orthostatic symptoms, depression, anxiety, or hyperechogenicity on substantia nigra ultrasound.
In relation to possible trials of potentially neuroprotective agents, this large study is helpful in guiding choice of stratification factors based on relative risk, and also in estimating the sample size needed – which the authors put at 142 to 366 patients per arm.25
Also highly relevant to the design of future trials of prevention is the Michael J Fox Foundation’s groundbreaking Parkinson’s Progression Markers Initiative. As of January 2019, this had completed enrollment of cohorts of people at enhanced genetic risk of PD, those in the PD prodrome, those with newly diagnosed but untreated PD, those with SWEDD (motor symptoms suggestive of PD but scans without evidence of dopaminergic deficit), and healthy controls.26 In total, longitudinal biomarker data will be available on more than 1400 people. Measures include motor function; NMS such as olfaction, RBD and cognition; MRI, PET and SPECT imaging; and the serial collection of biofluid samples from blood and CSF for assay of α-synuclein.
In the high-risk and prodromal cohorts, it is hoped that identifying a symptom and biomarker signature highly predictive of conversion to PD will open a window of opportunity for early intervention with neuroprotective strategies.
Access collection: Non-motor Symptoms in Parkinson’s Disease
Related content

The Braak staging scheme is a valuable model for conceptualizing PD progression and categorizes PD into stages of neuropathological changes.

This infographic provides a visual, at-a-glance overview of the Non-motor symptoms in Parkinson’s Disease.

In this documentary film, developed by the team behind The Brain Prize at the Lundbeck Foundation, you can learn more about how we mammalians move and the research that led to The Brain Prize in 2022.