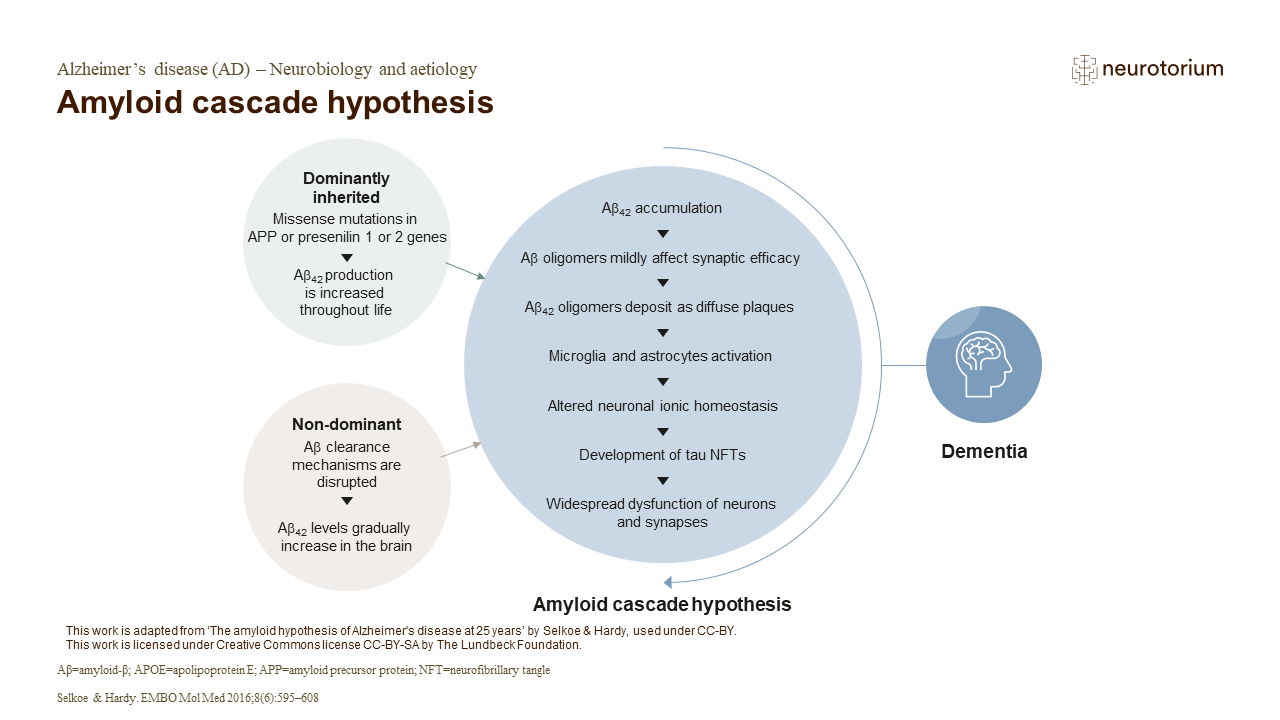
The amyloid cascade hypothesis suggests that Aß deposition is the causative agent in AD, while the tau neurofibrillary tangles, vascular damage, cell loss, and dementia occur as a direct result of the Aß deposition.2 Accumulation of Aß42 in limbic and associated cortices leads to gradual deposition of Aß42 oligomers as diffuse plaques.1 This then triggers an attendant inflammatory response following microglial and astrocytic activation, eventually leading to the development of tau NFTs caused by altered kinase/phosphatase activity. 1 As a result of these changes, widespread neuronal dysfunction, including selective neuronal loss and attendant neurotransmitter deficits, occur.1
The strongest data to support this idea comes from human genetics; however, preclinical cell culture and animal studies also support this hypothesis.1 Primary rat neuronal cultures exposed to aggregated Aß species demonstrate tau phosphorylation, neuritic damage, and dendritic spine loss.3 Discrepancy between the chronological appearance of amyloid plaques, tau neurofibrillary tangles, neuronal loss, and clinical dementia has caused controversy with the amyloid hypothesis.4 Many humans exhibit abundant Aß deposits at old age or death, without noticeable symptoms.1,4 The time between appearance of plaques and onset of tau pathology may be several years, with onset of clinical dementia following decades later, and therefore it is suggested that amyloid plaque pathology represents preclinical AD.5.6 To successfully delay the progression of the Aß cascade, multiple therapies targeting several aspects of the neurodegenerative network may be required, and the timing of the therapies during AD progression will be crucial for beneficial outcomes.2
References:
1.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016; 8 (6): 595–608.