

Why is genetics relevant and important?
The journey of understanding the role of genetics in PD began with the discovery of SNCA and PRKN in 1997 and 1998, respectively (see Table 1 for a timeline of developments in the PD genetics field). Mutations in these genes were found to be responsible for causing autosomal dominant early-onset PD (EOPD) (i.e., onset age <50 years) in the case of SNCA,5 and autosomal recessive juvenile-onset PD (<21 years) in the case of PRKN.6 These cases clearly put to bed a prevailing notion at the time that PD was “acquired” with genetics playing “no significant role in the aetiology of PD”.7,8
These, and subsequent findings in other monogenic and complex/sporadic forms of PD, also spurred major strides in understanding the molecular underpinnings of the disease.9 For example, aggregation of the a-synuclein protein (encoded by SNCA) and defects in mitochondrial function (with which PRKN is involved), are widely understood to be key players in the pathogenesis of the disease.10,11 There are now eight genes that have been implicated in monogenic PD (mutations in SNCA, LRRK2, GBA1, RAB32, and probably CHCHD2 causing autosomal dominant PD; and PRKN, PINK1 and DJ-1 causing autosomal recessive PD), and several more cause parkinsonian syndromes more generally.3,12 Several of these molecular pathways are currently being targeted in clinical trials (readers are referred to the Table in Lim et al. 20243 for an up-to-date summary of the gene targets and studies).
Importantly, there seems to be a convergence of the molecular pathways involved in both monogenic (or “Mendelian”) forms of PD (i.e., where the cause of PD is attributed to relatively rare mutations in a single gene) and the more common “idiopathic” (or “complex”) form,13 which occurs sporadically later in life. In the latter scenario, a complex interplay of genetic, environmental, ageing, and other factors is believed to underlie disease development. It is therefore hoped that genetics-informed therapies found to be effective in the smaller subset of monogenic PD patients can also be successfully applied in sporadic PD, at least in some cases. For example, it is anticipated that sporadic PD patients found to have elevated LRRK2 kinase activity (which is found in most, if not all, forms of monogenic LRRK2-related PD)14 could also be treated with LRRK2 kinase inhibitors.
Genetics also lends itself very well to the recent paradigmatic shift in the field towards a biological classification and definition of the disease,15-17 allowing patients, including those with fully or highly penetrant monogenic forms, to be diagnosed or classified and potentially recruited into clinical trials during an early window, before the disease process has become too advanced to be salvageable using disease-modifying therapies.18
Genotype-phenotype correlations
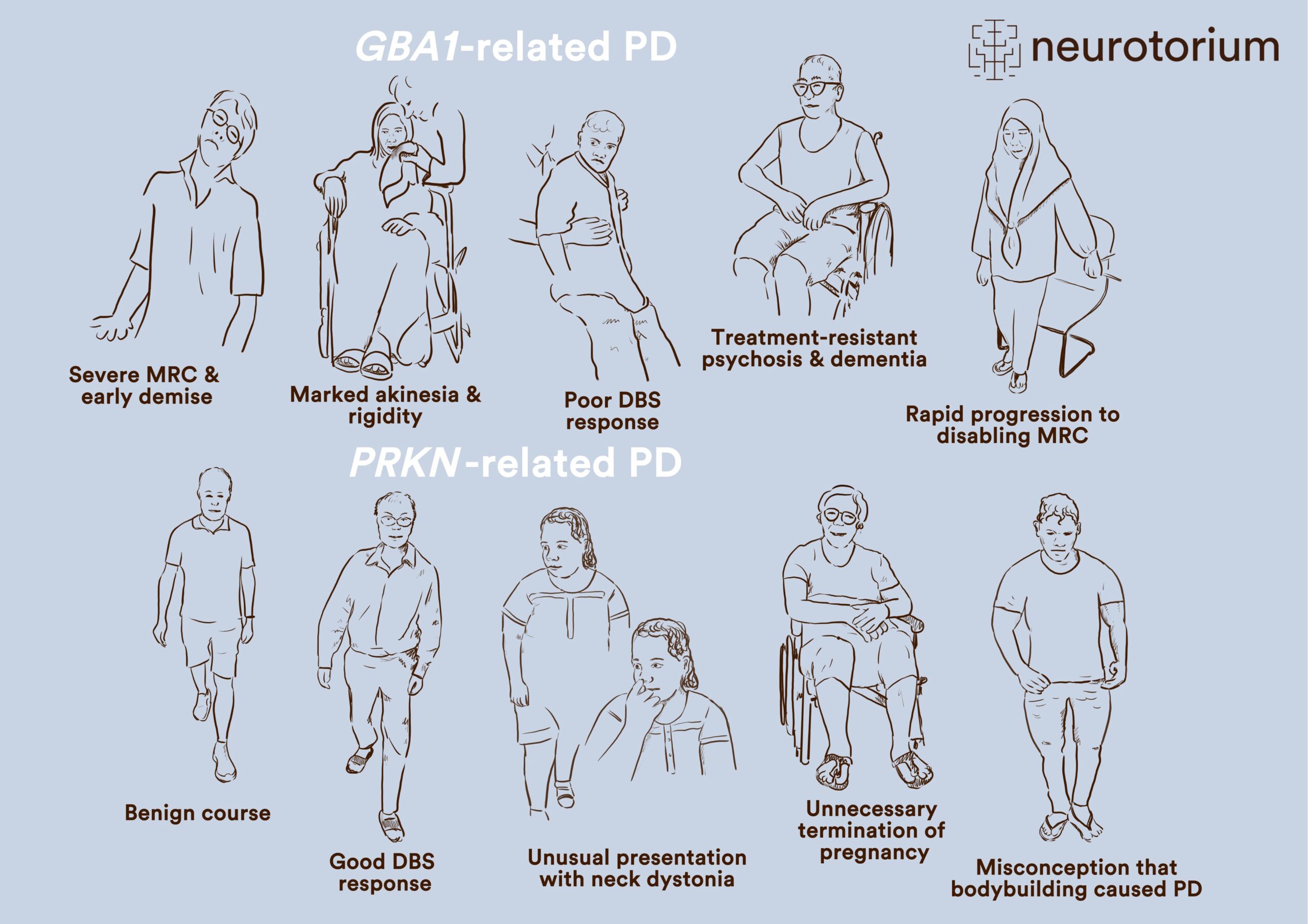
Besides being a springboard for understanding disease mechanisms, ultimately leading to the deployment of novel molecularly-targeted therapies, genetic information is also increasingly relevant in managing patients in the clinic. Some examples are depicted in Figure 1 and Slide 1.
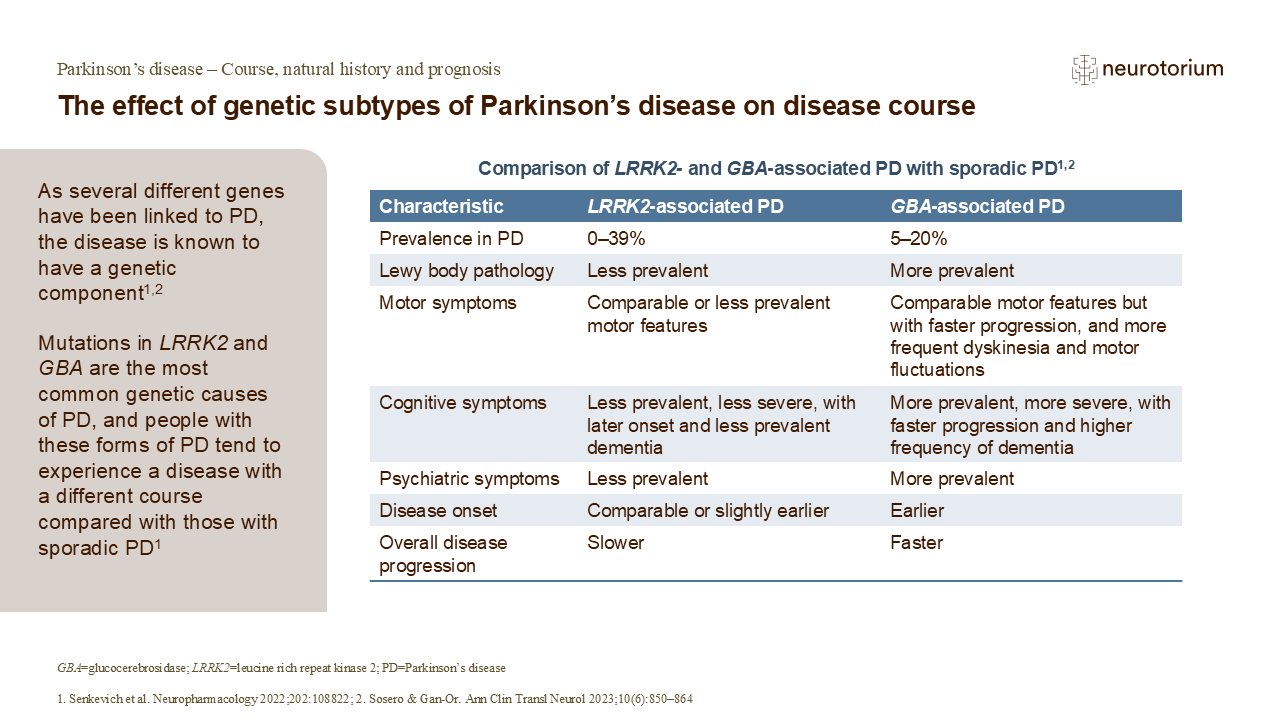
Thus, in the case of patients harbouring pathogenic GBA1 variants, clinicians are better able to understand why some patients demonstrate an aggressive clinical course, including the development of early motor response complications, “axial” motor features (such as speech and swallowing difficulties, as well as gait and balance problems), dementia and psychosis, and mortality.19-21 This can be observed even in patients with EOPD, who conventionally have been considered to experience a more slowly progressive disease course.22 There is some evidence suggesting that the more rapid disease progression is in part related to a heavier burden of synucleinopathy/Lewy pathology occurring in these patients (as well as in those harbouring SNCA mutations).19

Figure 1. GBA1- and PRKN-related PD exhibit multifaceted issues in patients
Multifaceted issues faced by patients with Parkinson’s disease (PD) and how genetics knowledge, using the examples of GBA1– and PRKN-related PD, can be applied clinically to aid personalized management. This includes a better understanding of the disease course and prognostication; enhanced selection and informed decision-making for treatments like deep brain stimulation (DBS); improved diagnosis in patients with atypical clinical features; assisting in family planning; and allaying anxiety, fear, or guilt. These early-onset PD patients were all seen by a movement disorders neurologist (SYL), underscoring the fact that these forms of PD are not rare. Moreover, the ancestries of these patients (all Chinese, Indian or Malay) are highly relevant globally, since China and India are by far the two most populous countries in the world, home to ~2.9 billion people (not counting the extensive diaspora of Chinese and Indians globally), and Malays comprise ~200 million individuals living in Southeast Asia.
Another important genotype-phenotype correlation is that some patients with GBA1 variants may not show a good response to deep brain stimulation (DBS) therapy,23,24 although others do seem to respond well.25 Studies have shown a relatively high frequency of GBA1 variant carriers among patients presenting for and undergoing DBS, which is perhaps unsurprising since many of these patients have troublesome motor response complications (the primary indication for DBS) and are younger (whereas older age, particularly >70 years, is a relative contra-indication for DBS).24
On the other hand, generally good long-term outcomes are seen with PRKN-related PD.20,26 In these patients, PD motor symptoms progress more slowly, and motor response complications only develop many years after disease onset27. Dementia is uncommon and survival is prolonged.20,26,27 Even so, the concepts of PRKN-related PD being a relatively “pure nigropathy” without significant extra-nigral neuropathology, and without synucleinopathy, have been challenged more recently with the observations that a substantial proportion of patients do manifest autonomic dysfunction,27 and test positive on a-synuclein seed amplification assays.28
Patients with PRKN-related PD typically respond well to DBS, knowledge of which is useful for patient selection and counselling. Because patients often present at a very young age and sometimes with unusual clinical features (such as focal dystonias or prolonged isolated tremor), the correct diagnosis can be delayed by years or even decades, with patients missing out on appropriate treatment and sometimes being labelled as having functional neurological disorders.29,30 A proper understanding of the recessive nature of the disease can also be very helpful in family planning (e.g., the disease being very unlikely to manifest clinically in offspring), and may avoid unfortunate situations like termination of pregnancy because of an erroneous assumption that a very early onset of PD portends a similar affliction in the offspring.31 Feelings of guilt in some patients who think their PD is caused by something they did – not an uncommon belief among patients and caregivers32 – can sometimes be allayed by being informed about the real (genetic) basis of their condition.
Patients with PRKN-related PD typically respond well to DBS, knowledge of which is useful for patient selection and counselling.
The genetic epidemiology of PD
Recognized monogenic forms currently comprise a minority of PD cases, perhaps around 5-15% globally.2,3 However, in selected populations, this figure can be considerably higher, due to factors such as founder effects (e.g., in the case of the LRRK2 p.G2019S variant, believed to have arisen in an ancient Middle Eastern founder) or high rates of consanguinity (e.g., in “MENASA” region [Middle Eastern, North African, and South Asian] countries).3 Furthermore, a very large number of familial cases, having for example what appear to be autosomal dominant or autosomal recessive patterns of transmission, currently remain “unsolved”.8 Technological advances such as long-read sequencing (LRS) and optical genome mapping (OGM)33 are starting to make a dent in these “cryptogenic” cases.34 Over time, together with newly discovered genes35,36, these and other developments will increase the number of cases that can be attributed to defects in single genes.
Recognized monogenic forms currently comprise a minority of PD cases, perhaps around 5-15% globally2,3
Major strides are also being made in understanding the genetic underpinnings of “idiopathic” or “complex” PD, facilitated especially by the increasingly large sample sizes of genome-wide association studies (GWAS), and fine-mapping enabled by the inclusion of ancestrally-diverse populations.37 Further development in these areas is expected to yield polygenic scoring (PGS) models that can, especially when combined with other (e.g., clinico-demographic and molecular markers) data, accurately predict not only disease development, but also a variety of important variables and outcomes such as age at disease onset, motor progression, the development of cognitive impairment with disease progression or impulsive-compulsive behaviours triggered by dopamine agonist treatment, and responsivity to medications and surgical therapies (pharmacogenomics and surgicogenomics).8,38-40
Concluding remarks
In all these efforts, the scientific and ethical imperatives of being inclusive of populations that have hitherto been under-represented in genetics research, are increasingly acknowledged.3,41,42
Major initiatives such as the Global Parkinson’s Genetics Program (GP2) have been very active in promoting not only the collection of genetic samples and data from around the world43, thereby facilitating access to genetic testing for patients and clinicians who otherwise may not have access to this,44,45 but also, from its inception, emphasizing local and regional capacity-building. This is done via numerous educational initiatives, and by funding training and clinical and research infrastructure in underserved areas.46 Moving forwards, it will be critical that all stakeholders strive to ensure that the fruits of genetics research, including access to clinical trials and new therapies, are shared equitably among patients and families worldwide, and not inadvertently contribute further to healthcare disparities between the haves and the have-nots.3,47
Moving forwards, it will be critical that all stakeholders strive to ensure that the fruits of genetics research, including access to clinical trials and new therapies, are shared equitably among patients and families worldwide
| Monogenic | Year | GWAS | Total Number of PD Participants | Ancestral Diversity |
| SNCA5 | 1997 | – | – | European |
| PRKN6 | 1998 | – | – | East Asian |
| PARK7/DJ-148 | 2003 | – | – | European |
| SNCA triplication49 | 2003 | – | – | European |
| PINK150 | 2004 | – | – | European |
| SNCA duplication51 | 2004 | – | – | European |
| LRRK252,53 | 2004 | – | – | European |
| 2005 | 1 risk variant, 2 tagged loci54 | 0.8k | European | |
| 2006 | 1st stage analysis55 | 0.3k | European | |
| GBA1 association56,57 | 2009 | – | – | Multiple |
| 2009 | SNCA, MAPT, LRRK2, PARK1658 | 2.0k | European | |
| 2009 | SNCA, MAPT, LRRK2, PARK16, BST159 | 2.0k | East Asian | |
| VPS3560,61 | 2011 | – | – | European |
| 2011 | 11 loci62 | 12.4k | European | |
| 2011 | 16 loci63 | 15.8k | European | |
| 2014 | 24 loci64 | 19.1k | European | |
| 2017 | 3 loci65 | 5.1k | East Asian | |
| 2017 | 44 loci66 | 26.0k | European | |
| 2019 | 6 loci67 | 28.6k | European | |
| 2019 | 11 loci68 | 4.1k | European | |
| 2019 | 78 loci (90 risk signals)69 | 37.7k | European | |
| 2020 | 11 loci70 | 6.7k | East Asian | |
| 2021 | XWAS: 2 loci71 | 11.1k | European | |
| 2021 | 1 variant East Asian specific, 1 variant shared72 | 40.2k | Multiple | |
| 2021 | 1 locus73 | 0.8k | Latino | |
| 2022 | 2 loci74 | 8.5k | Multiple | |
| 2023 | STR analysis: 34 loci75 | 16.6k | European | |
| 2023 | GWAS & CNV association study: 23 SNV, 1 CNV loci76 | 0.4k | East Asian | |
| 2023 | 8 loci77 | 2.0k | East Asian | |
| 2023 | 3 loci78 | 2.6k | European | |
| 2023 | WGS & WES: Rare variant burden tests: 2 loci79 | 7.2k | European | |
| 2023 | 1 risk variant80 | 1.5k | African | |
| 2023 | 78 loci37 | 49.0k | Multiple | |
| RAB32 p.S71R{{35} | 2024 | – | – | Multiple |
Table 1. A brief historical timeline of Parkinson’s disease (PD) genetics and genomics research
The discovery of monogenic causes of PD and GBA1-PD as well as their respective years of discovery are shown. Genome-wide association studies (GWASs) and brief overviews of their findings are also shown with the corresponding years in which they were published. The GWAS studies have over time increased in size, ancestral diversity, resolution, and scope. Adapted from Reference 3, Appendix Figure 1.
CNV: copy number variation; GWAS: genome-wide association study; k: thousand (rounded to the closest 100); PD: Parkinson’s disease; SNV: Single nucleotide variant; STR: Short-tandem repeat; WES: Whole exome sequencing; WGS: Whole genome sequencing; XWAS: chromosome X-wide association study.
Related content

The basal ganglia regulate movement through dopaminergic input from the SNc to the striatum, modulating motor signals via direct and indirect pathways.

Dopamine modulates movement by acting on D1 and D2 receptors in the striatum, influencing excitatory and inhibitory motor pathways.

{kind=link}
α-synuclein plays a central role in Parkinson’s disease, progressing from monomers to fibrils and Lewy bodies — the pathological hallmark of PD.