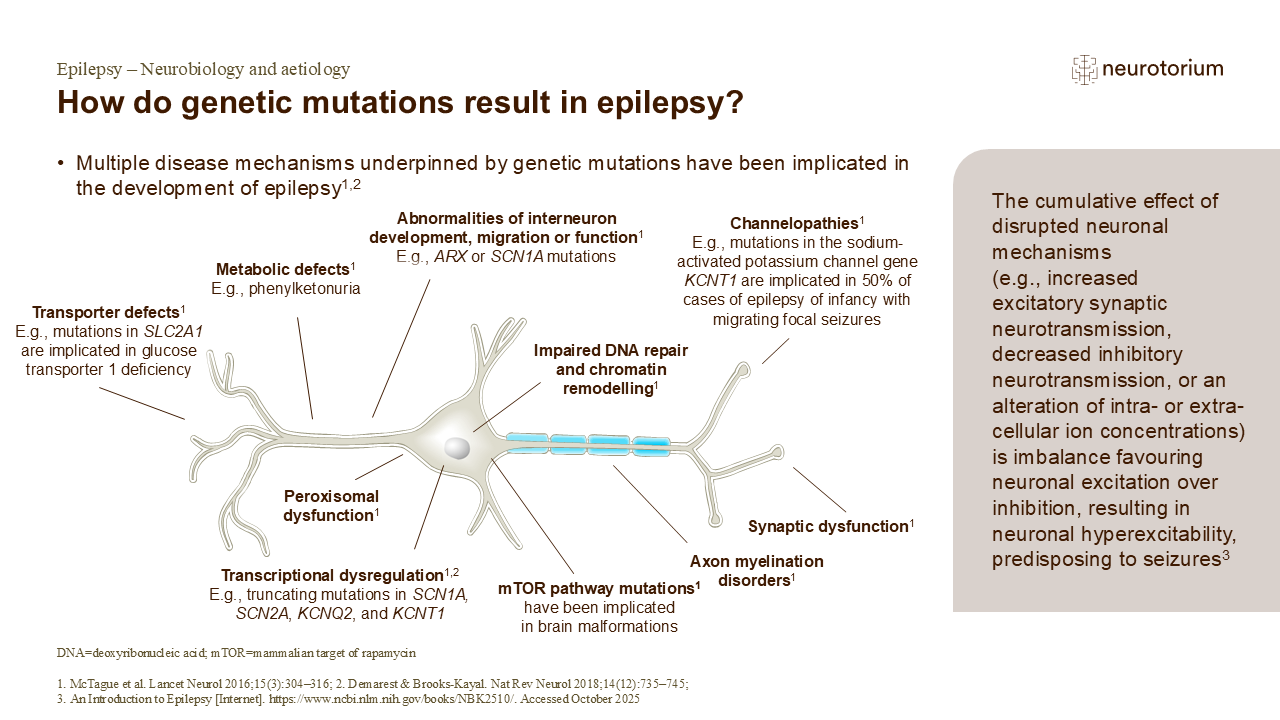
Genetic mutations can be passed down from a parent, or they can arise spontaneously in the individual.1 They result in an alteration – loss or gain – of the function of the affected gene or of the protein encoded by the gene, and subsequently in a disruption of one or more of the biological processes underpinning normal neuronal and brain function, associated with epilepsy.1 Loss-of-function mutations in sodium channel gene SCN1A have been detected in most patients with Dravet syndrome and gain-of-function mutations in the sodium-activated potassium channel gene KCNT1 have been detected in around half of infancy epilepsy cases with migrating focal seizures.1
Genetic causes of epilepsy can also affect the function of neurons through mechanisms unrelated to seizures, e.g., aberrant neuronal migration or the formation of abnormal neuronal networks.1
Variation in phenotype of genetic epilepsy can be driven by changes at the gene level, within the neuron, or across networks of neurons.2
References: